Has Quantum Computing Really Achieved Molecular Simulation?
In one sentence: they used a quantum computer to derive structure parameters from NMR spectra.

Recently, many tech outlets have claimed that Google’s Willow quantum chip can now “simulate molecular structures,” suggesting that drug and materials design are entering a “quantum era.” It sounds exciting—but it’s largely an over-interpretation of the original research.

To understand what this work actually achieved, we need to look at the source: “Quantum computation of molecular geometry via many-body nuclear spin echoes,” a paper by Google Quantum AI and UC Berkeley, published on arXiv.
Importantly, Google’s own announcement never claimed to have “solved molecular simulation” or “enabled drug design.”
What the Paper Really Did
This study doesn’t aim to “simulate all molecules.” Instead, it demonstrates how a quantum computer can help solve an NMR spectral inversion problem using the OTOC (Out-of-Time-Order Correlator) algorithm.
In simple terms, the researchers used quantum computing to help interpret complex nuclear magnetic resonance (NMR) signals—extracting structural parameters such as inter-atomic distances and torsion angles from real molecular data.
This is an exploratory work at the intersection of physical experiment, quantum information, and structural chemistry. It’s still far from “full molecular simulation,” but scientifically, it’s a meaningful first: applying NISQ-era quantum hardware to an actual molecular structure-learning task.
In one sentence: they used a quantum computer to analyze NMR spectra.
A Brief Refresher: NMR Structure Determination
Undergraduates in chemistry are often taught NMR structure determination through J-coupling constants—measuring coupling between two nuclei and using the Karplus equation to infer their torsion angle (φ):
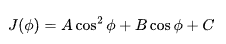
Knowing the J value allows one to estimate the spatial angle between atoms. This works well for small molecules and short-range couplings, typically through three bonds.
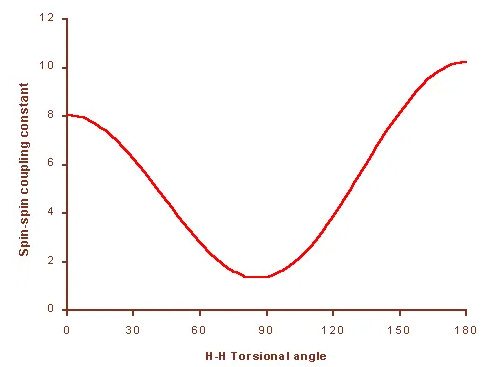
However, OTOC offers a new way: instead of measuring coupling, it measures how far an interference signal can travel. Experimentally, one excites a single nucleus and lets the signal propagate through the molecule. Then the system is “time-reversed” to see if the signal returns. Any disturbance from other spins will “scramble” it, and the strength of the echo reflects internal molecular geometry.
By fitting the OTOC decay curves, one can deduce hydrogen–hydrogen distances or torsion angles—even in cases too complex for traditional NMR methods.
Why Use Quantum Computers for OTOC?
Implementing OTOC requires both forward and reverse time evolution of the quantum system. Conceptually, you let the system evolve, reverse it, and measure how much information is lost. The loss reveals how far perturbations propagate within the system.
Classically, simulating this is extremely hard—it involves exponentially large Hilbert spaces and intricate interference effects. But for quantum hardware, this process is native language. Quantum bits inherently support superposition and interference, making them ideal for reproducing these dynamics directly, with far less computational overhead than reconstructing a full wavefunction.
That’s why OTOC is often considered one of the most practical near-term tasks for quantum computers.
What Exactly Did They Do in This Study?
The researchers focused on two molecular parameters:
The ortho–meta H–H distance in toluene, and
The torsion angle between rings in 3’,5’-dimethylbiphenyl.
They performed NMR experiments in a liquid-crystal environment to obtain the OTOC signal. Then, using classical molecular dynamics (OpenMM), they built structural models and treated the OTOC signal as the target function. Adjustable structural parameters were tuned until the simulated and experimental OTOC curves matched.

This approach is somewhat analogous to Reverse Monte Carlo (RMC)—iteratively adjusting force-field parameters and molecular geometries until the simulated spectrum fits the experimental one. The drawback is that such solutions may represent only local minima, rather than the true global structure.
In practice, if one just wants to interpret a simple NMR spectrum, it’s far more efficient to run a molecular dynamics simulation with a machine-learning force field—or even an ab initio molecular dynamics (AIMD) run—and analyze the resulting trajectories. Using DFT-derived potentials would be far more direct than bringing in a quantum computer.
In Summary
Google’s work does not represent a general solution to molecular simulation or drug design. However, it introduces an intriguing possibility: leveraging quantum computing to interpret complex multi-body signals in NMR experiments and extract structural information that’s otherwise inaccessible.
The Willow chip is therefore an important milestone—not for “quantum drug design,” but for demonstrating a realistic bridge between experimental chemistry and quantum information processing.
What’s Really Worth Watching in Quantum Chemistry
Rather than hyped claims, the most promising directions for quantum computing in chemistry are:
Redesigning core algorithms (e.g., quantum Monte Carlo) to natively suit quantum architectures—allowing computation of interaction energies, electron densities, and correlation effects.
Using quantum hardware to accelerate specific computational bottlenecks, such as numerical integration, Fourier transforms, fast multipole methods, matrix diagonalization, pair-interaction summations, or path-integral molecular dynamics.
Until then, the real revolution in drug and materials design still comes from deep learning, not quantum hardware.
