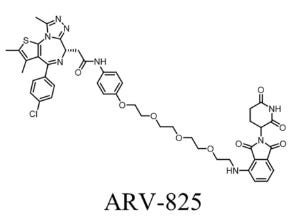
Unconventional Linker Design: From Radiopharmaceuticals to PROTACs

In modern drug discovery, linker design is moving from a supporting role to center stage. This is especially true in the development of radiopharmaceuticals and PROTACs (Proteolysis Targeting Chimeras), where the linker often determines not only whether the molecule works, but how well.
Traditionally, linker strategies fall into three broad categories:
Short, flexible linkers such as –CH₂–CH₂– or short PEG units, which provide basic spacing and mobility.
Long, flexible linkers such as PEGₙ or aliphatic chains, which create more distance between functional groups — a useful trick in radiopharmaceuticals to minimize radiation damage to the target binding site.
Rigid linkers, which restrict conformational freedom and help lock a molecule into the geometry needed for activity.
But in both radiopharmaceuticals and PROTACs, conventional approaches are beginning to run into limits. Researchers are now exploring unconventional linker design — guided not only by chemical intuition but also by AI-driven molecular generation.
A Quick Introduction to SMILES
The linker design method described here uses NVIDIA’s GenMol module. Since it requires SMILES input for the two terminal fragments of the linker, here’s a quick refresher.
SMILES (Simplified Molecular Input Line Entry System) is a way of representing molecular structures as text strings. For example:
Water: O
Ethanol: CCO
Benzene: c1ccccc1
Branches in SMILES are represented with parentheses (). For instance, CC(O)C represents a three-carbon chain where the middle carbon carries a hydroxyl branch.

SMILES can also represent multiple molecules within the same string. Different molecules are separated with a dot . to indicate coexistence without covalent bonds. For example, one benzene and two water molecules can be written as:
c1ccccc1.O.OAttachment points can be specified with markers such as [*1]. This notation indicates an atom in the molecule that can form a covalent bond with another fragment or linker. By marking such connection points, two molecules can be linked together with a specified linker to generate a new complete molecule.
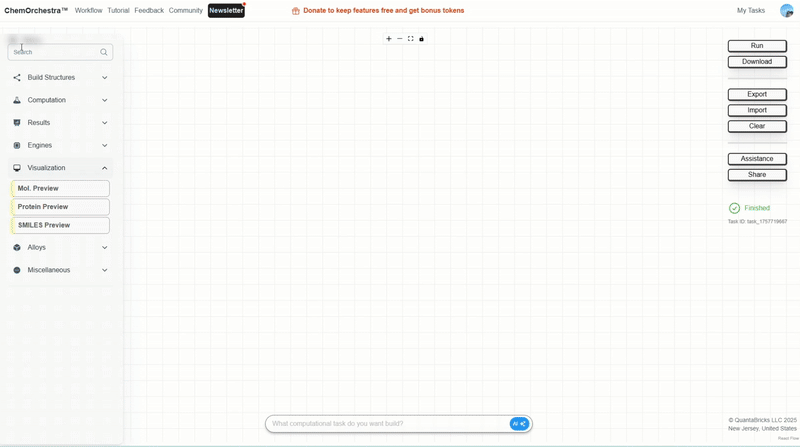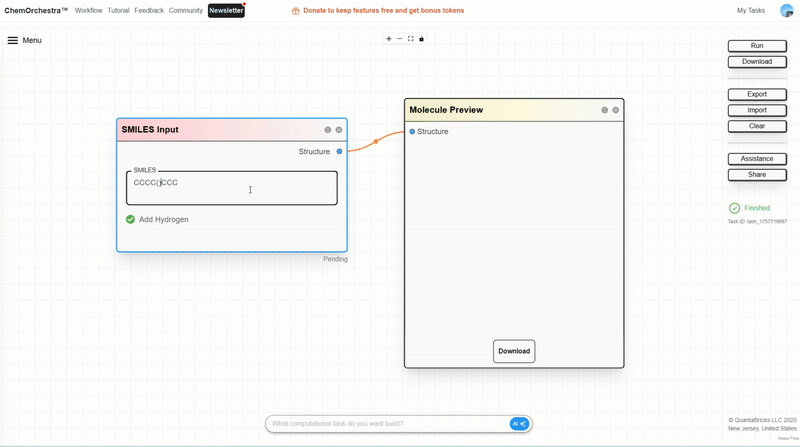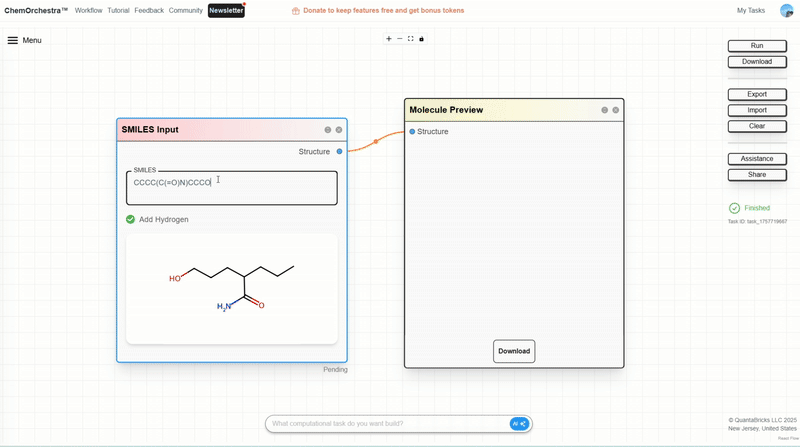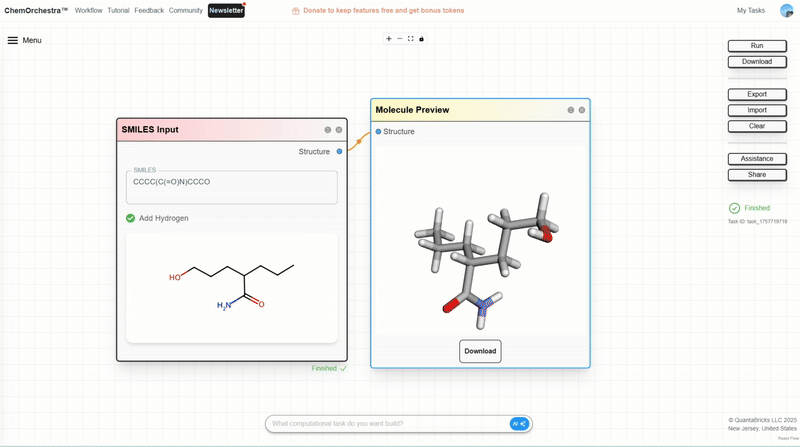
You can experiment with this in the SMILES Input module, which also converts SMILES strings into 3D molecular structures.
For more SMILES tutorials, see: Wikipedia: SMILES
Designing Molecules with GenMol
GenMol is an AI molecule generator based on a diffusion model developed by NVIDIA. It produces chemically reasonable structures with drug-like properties.
Step 1: Build a molecular generation workflow. From Build Structure, drag in GenMol, and from Visualization, drag in SMILES Preview. Connect them.
Step 2: Configure the GenMol module and choose Linker_design.
Step 3: Input the two terminal fragments to be connected by the linker. Join their SMILES strings with a dot . and specify the connection sites.
Example: linking a radioactive iodine-containing fragment with acetylsalicylic acid:
O=C(C([2*]))Oc1ccccc1C(=O)O.CNc1c(I)nc(O)c([1*])c1

Step 4: In the right-side control panel, click Run and wait for results.
When you find a linker you like, record the SMILES string.

Full workflow setup:

Current Challenges
While the workflow is powerful, it has some practical limitations:
SMILES editing isn’t intuitive: many chemists prefer drawing in ChemDraw. Planned updates will add drawing modules and image-to-SMILES conversion.
No direct linker length control: length can be tuned, but only by pairing GenMol with additional tools.
Compute cost: GenMol consumes GPU tokens. Free users get ~30–50 molecules from 5 tokens; additional tokens or VIP subscriptions unlock higher-end nodes like Boltz-2 or RFdiffusion. Boltz-2 shows 70+% accuracy in binding pose prediction.
Summary & Discussion
Some people have asked me how to design PROTAC linkers, but I’m still unsure about the broader scientific or industrial significance of unconventional linkers. Perhaps with AI and chemists’ intuition, better and more controllable linkers can be developed.
The best AI-driven linker design approach is to:
Generate thousands of PROTAC or radiopharmaceutical structures with length constraints.
Screen based on properties (rigidity, polarity, etc.).
Perform molecular docking and free energy calculations.
Select promising candidates for lab synthesis.
This is a niche need, and for most users I recommend working through a CRO to build a dedicated linker design platform, which can also be deployed locally. This tends to be cheaper than directly paying for ChemOrchestra compute time.
If you’d like to implement other molecular simulation features, contact us. We can demonstrate—via video—how to set up your own simulation workflows.
Workflow Demo:
Packaged GenMol for Self-Deployment:
ChemOrchestra Main Site:
https://www.quantabricks.xyz
